-
Modelling IR Spectral Dynamics upon Symmetry Breaking of a Photo-Excited Quadrupolar Dye
A.E. Nazarov, A.I. Ivanov and E. Vauthey
Journal of Physical Chemistry C, 124 (2020), p2357-2369


DOI:10.1021/acs.jpcc.9b10565 | unige:132980 | Abstract | Article HTML | Article PDF | Supporting Info
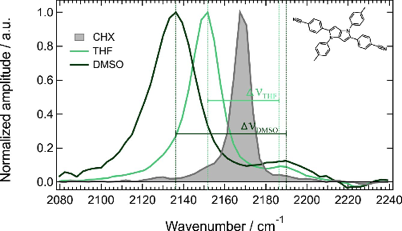
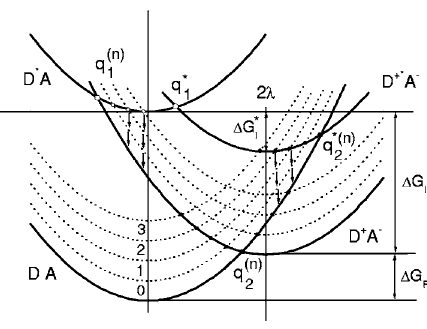
A significant number of quadrupolar dyes with a D-Ãâ¬-A-Ãâ¬-D oràA-Ãâ¬-D-Ãâ¬-Aàstructure, where D and A are electron donor and acceptor groups, were shown to undergo symmetry breaking (SB) upon optical excitation. During this process, electronic excitation, originally distributed evenly over the molecule, concentrates on one DâÃâ ââ¬â¢Ãââ¬Ã¢ââ‰â¬ÅA branch, and the molecule becomes dipolar. This process can be monitored by time-resolved infrared spectroscopy and causes significant spectral dynamics. A theoretical model of excited-state SB developed earlier (Ivanov, A. I. J. Phys. Chem. C2018,122, 29165âââ‰â¬Å29172) is extended to account for the temporal changes taking place in the IR spectrum upon SB. This model can reproduce the IR spectral dynamics observed in the -Cââ°Â¡C- stretching region with a D-Ãâ¬-A-Ãâ¬-D dye in two polar solvents using a single set of molecular parameters. This approach allows estimating the degree of asymmetry of the excited state in different solvents and its change during SB. Additionally, the relative contribution of different mechanisms responsible for the splitting of the symmetric and antisymmetric -Cââ°Â¡C-àstretching bands, which are both IR active upon SB, can be determined.